Can Stem Cells Treat Type 1 Diabetes? Exploring the Potential for Cellular Regeneration
Stem cell therapy has emerged as a promising area of research for treating Type 1 diabetes, which is...
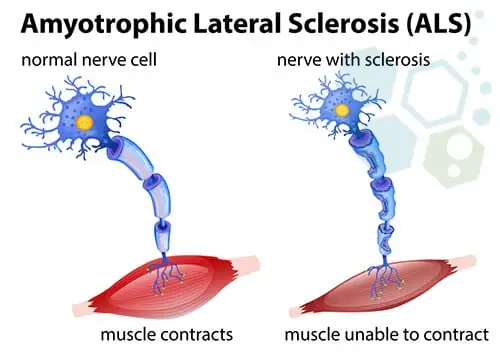
ALS, or Amyotrophic Lateral Sclerosis, is a progressive nervous system disease that affects the brain and spinal cord, leading to muscle weakness, loss of motor function, and ultimately, an inability to control essential body movements. Often known as Lou Gehrig's disease, after the famed baseball player who was diagnosed with it, ALS poses a significant challenge due to its complex and degenerative nature. While researchers have made strides in understanding ALS, there is currently no cure for this debilitating disease (Brown & Al-Chalabi, 2017).
ALS, short for Amyotrophic Lateral Sclerosis, affects motor neurons—nerve cells responsible for voluntary muscle movement, such as walking, speaking, and swallowing. Over time, ALS causes these neurons to gradually deteriorate and die, resulting in muscle atrophy and loss of voluntary motor control. The disease can impact any part of the body but commonly begins with symptoms in the arms or legs. It is a progressive condition, meaning symptoms worsen over time, affecting a person's ability to perform basic tasks (Mehta et al., 2023).
ALS symptoms vary from person to person, depending on the specific nerve cells involved. Some early signs and symptoms include:
ALS typically starts in localized areas, such as the hands, feet, or limbs, and then gradually spreads to other parts of the body. Muscle weakness becomes more severe as nerve cells continue to die, ultimately affecting chewing, speaking, swallowing, and breathing.

Source: Science Direct
The exact cause of ALS remains unknown, although some cases, about 10%, are hereditary. Scientists believe ALS may result from a combination of genetic and environmental factors (Hardiman et al., 2017). Here are some factors that can increase ALS risk:
As ALS progresses, it leads to several significant complications:
Although ALS has no cure, several treatments and interventions can improve quality of life. Respiratory assistance devices, feeding tubes, and communication tools can make life more manageable for those affected by the disease. Additionally, ALS treatment involves a multidisciplinary approach, including physical therapy, occupational therapy, speech therapy, and nutritional support .
Understanding the seven stages of ALS is crucial for patients, caregivers, and healthcare providers as they navigate each phase of this life-altering condition.
Key Takeaways:
ALS progresses through seven stages, each representing a different phase of motor neuron deterioration and its effects on patients' physical abilities and quality of life. Below is an in-depth overview of each stage, along with essential insights and supportive care strategies.

The initial phase of ALS often begins with subtle, mild symptoms. Muscle weakness, cramping, twitching (fasciculations), or stiffness may appear in various body areas, including the arms, legs, shoulders, or even the tongue. These symptoms are often overlooked, making early diagnosis difficult (National Institute of Neurological Disorders and Stroke, 2023).
As ALS advances, the motor neurons continue to weaken and atrophy, spreading to other muscle groups. Activities like walking, swallowing, and speaking gradually become more challenging (ALS Association, 2023). Adaptive tools and support from healthcare providers can help maintain daily functionality as much as possible during this stage.
In this stage, patients experience significant muscle weakness, often requiring mobility aids or assistance with daily tasks. Breathing and swallowing difficulties become more prevalent, highlighting the need for respiratory support and possibly alternative feeding methods (Zarei et al., 2015).
The fourth stage marks a rapid decline in physical abilities, necessitating around-the-clock care. Paralysis may extend to the limbs and trunk, and breathing and swallowing issues intensify (Taylor et al., 2016). Family members or caregivers must be prepared for increased dependency and the use of equipment such as ventilators to support basic functions.
During this stage, widespread paralysis impacts the arms, legs, and core muscles, leaving patients unable to speak or swallow independently. Ventilators and feeding tubes become essential for sustaining life and comfort, while the focus often shifts to palliative care and ensuring the patient's comfort (Kiernan et al., 2011).
At the end stage, ALS patients experience complete paralysis, with only minor eye movements remaining. Patients depend entirely on ventilators and feeding tubes, and communication methods, like eye-tracking software, may still provide a form of interaction (Brown et al., 2017).
The final phase of ALS involves total paralysis and full dependency on supportive care for basic life functions. Respiratory failure is often the cause of death, with the average survival rate from symptom onset being 3-5 years, though about 10% of patients live beyond a decade (Kiernan et al., 2011). End-of-life care prioritizes comfort, addressing pain, and supporting the family.
In the initial stages, symptoms of ALS may be subtle and vary greatly from one individual to another. Recognizing these early signs is essential for timely intervention and effective management.
| Symptoms | Description | Limb Onset Prevalence | Bulbar Onset Prevalence |
| Muscle Weakness | Difficulty with routine tasks | 60-70% | 20-30% |
| Muscle Spasticity | Muscle stiffness | Common | Less Common |
| Cramping | Painful muscle cramps | Often | Occasionally |
| Fasciculations | Muscle twitching | Often | Occasionally |
In the early stages, ALS can interfere with daily activities in subtle ways that progress over time:
Diagnosing ALS involves a careful process since no definitive test exists. A correct diagnosis is vital for appropriate care planning.
| Test | Purpose | Description |
| Blood and Urine Tests | Rule out other conditions | Checks for markers to eliminate other diseases |
| EMG | Evaluate muscle electrical activity | Measures electrical signals from muscles |
| NCS | Assess nerve function | Tests the speed and strength of nerve signal transmission |
| MRI | Rule out other conditions | Uses magnetic fields to create detailed images of the brain and spinal cord |
Early diagnosis offers several advantages:
In the middle stages, ALS symptoms typically worsen, leading to more significant loss of muscle control and independence.
The progressive nature of ALS often brings emotional and psychological challenges. Support from resources and caregivers is crucial during this stage.
As swallowing becomes more challenging, a gastrostomy tube, or G-tube, may be introduced to help with nutrition and hydration.
| Aspect | Description |
| Procedure | Placement of a gastrostomy tube (G-tube) |
| Benefits | Ensures adequate nutrition, hydration, and administration of medications |
In the advanced stages of ALS, most voluntary muscles are paralyzed, severely affecting functions like breathing, speaking, and swallowing.
The final stages are marked by full paralysis, and patients require constant care. The focus shifts to providing comfort and maintaining dignity.
Medications can provide symptom relief, and support services focus on delivering a dignified, comfortable experience.
Amyotrophic Lateral Sclerosis (ALS) has long been an enigma for researchers, devastating patients with progressive muscle paralysis while offering few treatment options. However, recent breakthroughs in stem cell therapy for ALS present new hope. Investigators are using stem cells both to treat and model ALS, with pioneering efforts like those at the Cedars-Sinai ALS Clinic pushing the boundaries of our understanding and potential treatments for this debilitating condition. This article delves into how stem cell therapy for ALS patients may revolutionize ALS treatment, and the ongoing research aiming to slow, stop, or even reverse the disease.

Stem cell therapy for ALS leverages the body's regenerative potential, using cells that can self-renew and differentiate into specialized cell types. For ALS, scientists are exploring neural progenitor cells that can differentiate into astrocytes—support cells for motor neurons that play a role in maintaining neural health. Studies show that using astrocytes modified to produce glial cell line-derived neurotrophic factor (GDNF), a potent growth factor that supports neuron survival, might offer protection to dying motor neurons (Svendsen et al., 2007).
In a breakthrough study published in Nature Medicine, Cedars-Sinai investigators demonstrated that it is safe to implant such engineered cells into the spinal cord of ALS patients (Chen et al., 2022). Moreover, options for stem cell therapy for ALS in Cayman Island have emerged, providing alternative avenues for patients seeking innovative treatments.
The Cedars-Sinai ALS Clinic team conducted an 18-patient trial in which neural progenitor cells were implanted into patients’ lumbar spinal cords, an area that controls leg movement. Patients acted as their own control, with only one side of the spinal cord receiving the stem cell treatment. This study marked a significant milestone by confirming the safety of this therapy, allowing further research into its efficacy (Svendsen et al., 2022).
Postmortem analysis revealed that the cells continued producing GDNF up to three and a half years after the procedure, providing potential long-term neuroprotective effects (Johnson et al., 2023). Investigations into adipose stem cell therapy for ALS are also ongoing, as researchers explore various stem cell sources to enhance treatment effectiveness.
In addition to treating ALS, stem cells are invaluable in research settings, helping scientists understand the cellular changes that lead to ALS. Cedars-Sinai scientists use induced pluripotent stem cells (iPSCs) derived from ALS patients to create disease models in petri dishes. These models mimic patient-specific disease characteristics and allow researchers to observe ALS progression in real-time (Ho et al., 2021). iPSCs offer the unique advantage of capturing genetic and molecular differences across patients, enabling researchers to explore the possibility that ALS might encompass a spectrum of disorders rather than a single disease.

Using patient-derived iPSCs, researchers have observed different molecular signatures in sporadic versus familial ALS cases, shedding light on potential genetic contributors that may otherwise go undetected. In a study published in Cell Systems, Ho et al. (2021) found distinct RNA and protein expressions between ALS subtypes, suggesting that personalized therapies targeting specific cellular dysfunctions may eventually be possible.
The future of ALS research may lie in personalized medicine, where treatments are tailored to the individual’s genetic and cellular makeup. With advancements in technology, researchers at Cedars-Sinai are classifying ALS based on genetic markers and cellular behaviors, which may lead to targeted interventions designed for specific ALS subtypes (Sareen et al., 2023). This approach is critical, as genetic factors appear to play a role in only a minority of ALS cases, with the vast majority presenting with sporadic forms of the disease that have no clear genetic cause.
The team at Cedars-Sinai has assembled the largest collection of ALS-specific iPSCs in partnership with the Answer ALS initiative, creating a repository of genetic, biochemical, and clinical data to fuel future discoveries (Answer ALS, 2023). Researchers worldwide can use this open-source resource to deepen their understanding of ALS mechanisms and explore novel treatment pathways.
Notably, the Shiney Wellness stem cell therapy for ALS patients has gained attention for its structured and comprehensive approach. Their protocols emphasize rigorous patient selection and follow-up, ensuring that patients receive the highest quality of care while exploring the potential of stem cell therapy interventions.


